eISSN 2444-7986
DOI: https://doi.org/10.14201/orl.32877
ORIGINAL ARTICLE
VARIANTS IN RIPOR2 GENE RELATED TO HEARING LOSS ARE INFREQUENT IN NORTHERN SPAIN
Las variantes en el gen RIPOR2 relacionadas con la hipoacusia son infrecuentes en el Norte de España
Aida VEIGA-ALONSO; Julia FERNÁNDEZ-ENSEÑAT  ; Andrea MARTÍNEZ-CAMERANO ; Rocío GONZÁLEZ-AGUADO ; Esther ONECHA-DE LA FUENTE ; Carmelo MORALES-ANGULO
; Andrea MARTÍNEZ-CAMERANO ; Rocío GONZÁLEZ-AGUADO ; Esther ONECHA-DE LA FUENTE ; Carmelo MORALES-ANGULO
Department of Otolaryngology, Marqués de Valdecilla University Hospital, Santander. Spain.
Correspondence: aida.veiga@scsalud.es
Reception date: February 9, 2025
Date of Acceptance: April 18, 2025
Publication date: May 22, 2025
Date of publication of the issue: pending publication
Conflict of interest: The authors declare no conflicts of interest
Images: The authors declare that the images have been obtained with the permission of the patients
Rights policy and self-archive: the self-archive of the post-print version (SHERPA / RoMEO) is allowed
License CC BY-NC-ND. Creative Commons Attribution-Noncommercial-NoDerivate Works 4.0 International
University of Salamanca. Its commercialization is subject to the permission of the publisher
SUMMARY: Introduction and objective: To assess the prevalence and clinical characterization of variants in the RIPOR2 gene among patients with bilateral sensorineural hearing loss (SNHL) of unknown etiology in northern Spain.
Method: This 7-year (2018–2024) observational, retrospective, and descriptive study involved patients with bilateral SNHL treated at a tertiary hospital. Genetic analysis was performed using next-generation sequencing (NGS), employing a gene panel for sensorineural hearing loss. The presence of RIPOR2 gene variants was specifically analyzed.
Results: Among the 381 patients, two variants in the RIPOR2 gene were identified in three individuals (0.8 %): c.1879G>A (p.Asp627Asn) and c.3193G>A (p.Ala1065Thr). The variant c.1879G>A was detected in two patients, while the c.3193G>A variant was found in only one patient who also carried other pathogenic variants associated with hearing loss. The three patients exhibited cookie-bite hearing loss, predominantly affecting the 1000, 2000, and 4000 Hz frequencies, respectively, without associated vestibular symptoms.
Conclusions: variants in the RIPOR2 gene are infrequent among patients with SNHL in northern Spain (Cantabria). Moreover, there is no conclusive evidence supporting their pathogenicity in cases of SNHL with unknown etiology. Further studies are needed to clarify their clinical significance.
KEYWORDS: sensorineural hearing loss; genetics; audiology; non-syndromic hearing loss; RIPOR2 gene.
RESUMEN: Introducción y objetivo: Evaluar la prevalencia y caracterización clínica de las variantes en el gen RIPOR2 en pacientes con hipoacusia neurosensorial (HNS) bilateral de etiología desconocida en el norte de España.
Método: Estudio observacional, retrospectivo y descriptivo realizado durante siete años (2018–2024), que incluyó pacientes con HNS bilateral atendidos en un hospital terciario. Se realizó un análisis genético mediante secuenciación de nueva generación (next-generation sequencing, NGS), utilizando un panel de genes para hipoacusia neurosensorial. Se analizó específicamente la presencia de variantes en el gen RIPOR2.
Resultados: De los 381 pacientes, se identificaron dos variantes en el gen RIPOR2 en tres individuos (0,8 %): c.1879G>A (p.Asp627Asn) y c.3193G>A (p.Ala1065Thr). La variante c.1879G>A se detectó en dos pacientes, mientras que la variante c.3193G>A se encontró en un solo paciente, quien además presentaba otras variantes patogénicas asociadas con la hipoacusia. Los tres pacientes mostraron una pérdida auditiva en forma de "cookie-bite", afectando predominantemente las frecuencias de 1000, 2000 y 4000 Hz, sin síntomas vestibulares asociados.
Conclusiones: Las variantes del gen RIPOR2 en pacientes con HNS en el norte de España (Cantabria) son poco frecuentes. Además, no existe evidencia concluyente que respalde su patogenicidad en casos de HNS de etiología desconocida. Se requieren más estudios para esclarecer su significado clínico.
PALABRAS CLAVE: hipoacusia neurosensorial; genética; audiología; hipoacusia no sindrómica; gen RIPOR2.
INTRODUCTION
The RHO family of GTPases plays a crucial role in cell growth, migration, and polarization [1]. Furthermore, the RHO family interacting cell polarization regulator (RIPOR, formerly known as FAM65B), particularly RIPOR1, RIPOR2, and RIPOR3, is a family of proteins that regulates RHO GTPases [1]. These proteins are expressed in a wide variety of tissues, including the ear, specifically in hair cells and stereocilia [2, 3]. The fundamental role of cochlear hair cells in converting sound-induced vibrations into electrical signals relies on the specialized bundle of stereocilia located at the apical surface of the hair cell [4, 5]. A loss of function caused by mutations in RIPOR2 gene is associated with hearing impairment [4-8]. Even in murine models, it has been demonstrated that RIPOR2 deficiency results in profound hearing loss [2]. The mutated gene may cause cilia (hair cells) to be less firmly embedded in the cochlea, ultimately impairing the mechanotransduction apparatus due to altered stereocilia [3]. In tissues outside the inner ear, RIPOR2 loss of function could potentially be compensated by RIPOR1 and RIPOR3 due to their redundant functions. However, gene expression analysis has shown that RNA levels of RIPOR1 and RIPOR3 are low in hair cells [9, 10].
Alterations in the RIPOR2 gene, which encodes this protein, have been linked to hearing loss subtypes DFNA21 and DFNB104 [1, 2, 7, 8]. Diaz-Horta et al. identified a splice-site mutation (c.102-1G>A) in the RIPOR2 gene that completely cosegregated with the phenotype in a large consanguineous Turkish family with recessive nonsyndromic hearing loss [2]. On the other hand, an in-frame deletion mutation (c.1696_1707del) in RIPOR2 gene has also been linked with autosomal dominant (AD) hearing loss [7]. Subsequent studies demonstrated a relatively high prevalence of this variant in the Netherlands (0.052 %), where it has been identified as the cause of adult-onset DFNA21 hearing loss [8].
Furthermore, it has recently been shown that RIPOR2-mediated autophagy dysfunction is central to aminoglycoside-induced hearing loss [11].
Mutations in other genes, such as COCH, which were initially found to be relatively common among patients with hearing loss in the Netherlands [12], have also been shown to be frequent in northern Spain [13]. This raises the hypothesis that variants in the RIPOR2 gene, relatively common in this population, may also be prevalent in northern Spain.
The aim of our study was to determine the prevalence of variants in the RIPOR2 gene among patients with bilateral sensorineural hearing loss (SNHL) of unknown etiology in northern Spain and to assess the clinical characteristics of the patients carrying these variants.
MATERIAL AND METHODS
An observational, retrospective, and descriptive study was conducted on all seemingly unrelated patients with bilateral SNHL of unknown origin who underwent genetic testing at the Otolaryngology Department of a tertiary hospital (Marqués de Valdecilla University Hospital, Santander) from January 2018 to December 2024. The criteria used by the clinician in the prior selection of patients for the genetic study included all the following:
1. Bilateral SNHL
2. Suspicion of a genetic origin of hearing loss based on one or more of the following:
- The presence of a family history of hearing loss.
- Characteristic hearing profile (bilateral U-shaped or ascending hearing losses).
- Hearing loss with onset in childhood or adolescence.
- Patients who underwent cochlear implantation.
- Patients with more severe bilateral SNHL than expected in the context of presbycusis [14].
3. Absence of other causes that would justify the hearing loss.
The age of onset of hearing loss, mode of onset (sudden, fluctuating, or progressive), involvement (symmetric or asymmetric), evolution (stable or progressive), and the presence of other otological symptoms accompanying the hearing loss were all recorded. The progression of hearing loss was calculated in patients who had more than one tonal audiometry test conducted at different time periods.
The degree of hearing loss on tonal audiometry (at the time of the genetic diagnosis) was classified according to the criteria of the American Speech-Language-Hearing Association [15], as slight (16-25 decibels [dB]), mild (26-40 dB), moderate (41-55 dB), moderately severe (56-70 dB), severe (71-90 dB), or profound (91 dB or greater). The configuration of hearing loss, as seen on audiometric analysis at the time of the first audiometry, was classified as sloping, flat, rising (low frequency), or midfrequency (cookie-bite) loss [16].
Results from supplementary tests such as auditory brainstem response (ABR), otoacoustic emissions (OAEs), vestibular tests, CT scans, and/or MRIs were also collected.
Genetic analysis: After obtaining genomic DNA from peripheral blood, a massive sequencing study (NGS) was carried out using a targeted custom gene panel associated with hearing loss [17] including the RIPOR2 gene according to a previously described technique [17].
The classification of the variants was performed in accordance with the recommendations of the ACMG and the expert specifications outlined in the ACMG/AMP variant interpretation guidelines for genetic hearing loss [18]. Based on these guidelines, variants were categorized as pathogenic, likely pathogenic, variant of unknown significance (VUS), likely benign (LB), or benign.
Statistical Analysis: A descriptive statistical analysis was performed on the studied variables.
Ethical considerations: The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national and institutional guidelines on human experimentation and with the Helsinki Declaration of 1975, as revised in 2008. The study was approved by the Ethics Committee for Research with Medicines and Health Products of Cantabria (CEIM), internal code: 2024.104 (Acta 8/2024 de 22/03/2024).
RESULTS
Among the 381 patients, two variants in the RIPOR2 gene were identified in three individuals (0.8 %): c.1879G>A (p.Asp627Asn) and c.3193G>A (p.Ala1065Thr). The variant c.1879G>A was detected in two patients, while the c.3193G>A variant was found in only one patient who also carried other pathogenic variants associated with hearing loss. The three patients exhibited cookie-bite hearing loss, predominantly affecting the 1000, 2000, and 4000 Hz frequencies, without associated vestibular symptoms (Figure 1). The demographic and clinical characteristics are summarized in Table 1. Patient 1 exhibited brainstem potentials consistent with cochlear involvement. Patients 2 and 3 presented an inheritance pattern compatible not only with AD traits but also with mitochondrial inheritance. In both cases, mitochondrial genetic mutations known to cause hearing loss were ruled out.
Other family members declined to undergo genetic and clinical studies.
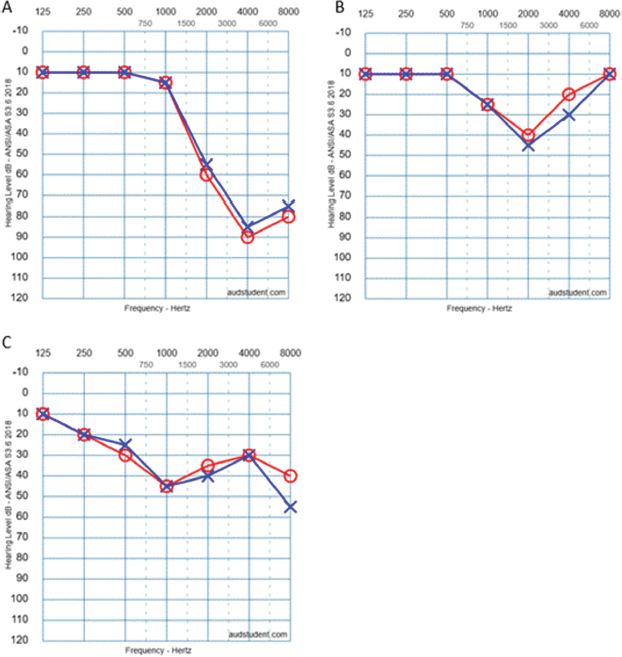
Figure 1. First audiogram performed for each patient carrying mutations in the RIPOR2 gene. A: Patient 1. B: Patient 2. C: Patient 3.
Table 1. Demographic, clinical, and genetic characteristics of patients with variant in the RIPOR2 gene.
Patient number/Age/Gender |
FH/inheritance pattern |
Age of onset |
Progression |
Audiometric profile |
Severity |
Vestibular Symptoms/Physical Examination/VT |
CT/MRI |
Genetic variant/Affected protein |
Zygosity |
Treatment |
P1/52/F |
UN |
<10 |
Yes |
High frequencies (especially 4000 Hz) |
Low frequencies: Normal Frequencies 4000, 6000 Hz: Severe |
No |
NR |
c.1879G>A, p.Asp627Asn |
Heterocygosis |
HA |
P2/57/M |
AD/Maternal |
30-40 |
Yes |
High frequencies (especially 2000, 4000 Hz) |
Low frequencies: Normal Other frequencies: Mild-moderate |
No |
NR |
c.1879G>A, p.Asp627Asn |
Heterocygosis |
No treatment |
P3/45/F |
AD/Maternal |
30-40 |
Yes |
Mid and high frequencies |
Mild to moderate at mid and high frequencies |
No |
NR |
c.3193G>A, p.Ala1065Thr Other variants in other genes: TMPRSS3, USH2A, TCM1, PDZD7 |
Heterocygosis |
HA |
P: proband. M: Male. F: Female. FH: Family history of hearing loss. UN: Unknown. AD: autosomal dominant. VT: Vestibular test. CT/MRI: computed tomography/magnetic resonance imaging HA: Hearing aids. NR: not realized.
DISCUSSION
The evidence linking homozygous pathogenic variants in RIPOR2 gene (DFNB104) is limited to a single study involving a Turkish family of six members segregating prelingual, profound, hearing loss [2] caused by a pathogenic variant in exon 3. This remains the only case reported in ClinVar as pathogenic or likely pathogenic.
For heterozygous variants (DFNA21), another study identified a heterozygous 12-nucleotide in-frame deletion (c.1696_1707del, p.Gln566_Lys569del) in RIPOR2 gene in 12 Dutch families with nonsyndromic hearing loss [7]. The presence of an identical variant in 12 families of shared ancestry, along with haplotype analysis, strongly suggests a founder effect [7].
In a subsequent study, researchers in the Netherlands analyzed the presence of this variant in a database of nearly 23,000 Dutch individuals who had undergone genetic testing [8]. They found that 18 individuals carried the c.1696_1707del variant. It is estimated that approximately 13,000 people in the Netherlands carry this defective gene [8]. Since hearing loss associated with this variant typically manifests later in life, it is estimated that around 9,000 individuals in the Netherlands are currently experiencing hearing loss caused by the variant [8]. This makes the RIPOR2 gene pathogenic variant one of the most common hereditary causes of hearing loss in the Netherlands [8]. However, it has not been described in other populations.
Notably, a large study conducted in Japan, including 10,047 patients, by Usami and colleagues did not identify any pathogenic mutations in RIPOR2 gene [19]. Similarly, no RIPOR2 mutations were detected in studies conducted in Iran [20], Spain [21-23], or broader European populations [24].
Genetic studies on hearing loss prevalence in Cantabria (Spain) have shown that the most frequently affected autosomal dominant genes are TECTA, KCNQ3, and COCH [17]. Our study demonstrates the very low frequency of RIPOR2 variants in this region of northern Spain (Cantabria). Furthermore, the pathogenicity of the two variants identified in our study remains uncertain.
The c.1879G>A variant, found in two unrelated probands in our cohort, was associated with a hearing loss pattern sparing low frequencies but presenting notches at 2000 and 4000 Hz. Given its very low frequency in population databases, it might represent a pathogenic variant. However, this could not be confirmed without family segregation studies, which were impossible to conduct due to the lack of cooperation from other family members.
As for the c.3193G>A variant, there was also a drop in cookybite at 1000 Hz, similar to the other variant. However, since it appeared alongside other variants in genes known to cause AD hearing loss, no conclusions can be drawn, as it might represent a benign variant.
The audiometric phenotype and age of onset of hearing loss associated with DFNA21 have been studied extensively only in cases with the RIPOR2 c.1696_1707del variant [7]. In total, this variant was detected in 59 out of 63 affected participants. The hearing loss associated with the variant is variable, and some carriers even exhibit normal hearing. However, as the average age of onset reported in these families is 30 (±14.9) years, with the latest onset recorded at 70 years, it is possible that some unaffected individuals might develop hearing loss in the future [7]. Nevertheless, incomplete penetrance of the variant cannot be ruled out [7]. The phenotypic variability likely results from an interplay between environmental and genetic modifying factors [7]. Interestingly, four affected individuals in the study who did not carry the variant were thought to represent phenocopies [7].
No vestibular or radiological abnormalities have been associated with pathogenic variants in RIPOR2 gene [2, 7].
There is a paucity of information regarding the treatment of patients with pathogenic RIPOR2 gene variants. Furthermore, there are no reports of cochlear implants in patients with mutations in the RIPOR2 gene. Seligman et al., in the largest published series of genetic studies in 459 cochlear implanted patients, didn´t identify pathogenic variants in the RIPOR2 gene [25].
Given the relatively high prevalence of pathogenic RIPOR2 gene variants linked to hearing loss in the Netherlands, efforts are underway in that country to develop gene therapy for DFNA21.
The limitations of our study include the small sample size and the inability to determine the pathogenicity of the two variants found, due to the lack of family segregation studies. Nonetheless, apart from the clinical and prevalence studies on RIPOR2 gene variants conducted in the Netherlands, ours is the only study exclusively addressing this aspect. We believe such studies are essential to elucidate the role of RIPOR2 gene variants in hearing loss of unknown origin.
CONCLUSIONS
Variants in RIPOR2 gene are infrequent among patients with SNHL in northern Spain (Cantabria), occurring in less than 1 % of patients with SNHL of unknown etiology. Additionally, we cannot conclusively establish the pathogenic nature of the variants identified.
REFERENCES
1.Lu Z, Ding Y, Cao W, Wang S, Gao K. Role of RHO family interacting cell polarization regulators (RIPORs) in health and disease: Recent advances and prospects. Int J Biol Sci. 2022;18(2):800-8. https://doi.org/10.7150/ijbs.65457
2.Diaz-Horta O, Subasioglu-Uzak A, Grati M, DeSmidt A, Foster J 2nd, Cao L, et al. FAM65B is a membrane-associated protein of hair cell stereocilia required for hearing. Proc Natl Acad Sci U S A. 2014;111(27):9864-8. https://doi.org/10.1073/pnas.1322632111.
3.Zhao B, Wu Z, Muller U. Murine Fam65b forms ring-like structures at the base of stereocilia critical for mechanosensory hair cell function. eLife. 2016;5:e14222. https://doi.org/10.7554/eLife.14222.
4.Goutman JD, Elgoyhen AB, Gomez-Casati ME. Cochlear hair cells: The sound-sensing machines. FEBS Lett. 2015;589(22):3354-61. https://doi.org/10.1016/j.febslet.2015.09.004.
5.Manor U, Kachar B. Dynamic length regulation of sensory stereocilia. Semin Cell Dev Biol. 2008;19(6):502-10. https://doi.org/10.1016/j.semcdb.2008.09.005.
6.Diaz-Horta O, Abad C, Cengiz FB, Bademci G, Blackwelder P, Walz K, et al. Ripor2 is involved in auditory hair cell stereociliary bundle structure and orientation. J Mol Med (Berl). 2018;96(11):1227-38. https://doi.org/10.1007/s00109-018-1704-6.
7.de Bruijn SE, Smits JJ, Liu C, Lanting CP, Beynon AJ, Blankevoort J, et al. A RIPOR2 in-frame deletion is a frequent and highly penetrant cause of adult-onset hearing loss. J Med Genet. 2020 Jul 6;57(12):861-7. https://doi.org/10.1136/jmedgenet-2020-106863.
8.Velde HM, Homans NC, Goedegebure A, Lanting CP, Pennings RJE, Kremer HJ. Analysis of Rotterdam Study cohorts confirms a previously identified RIPOR2 in-frame deletion as a prevalent genetic factor in phenotypically variable adult-onset hearing loss (DFNA21) in the Netherlands. J Med Genet. 2023;60(11):1061-6. https://doi.org/10.1136/jmg-2023-109146.
9.Mardakheh FK, Self A, Marshall CJ. Rho binding to FAM65A regulates Golgi reorientation during cell migration. J Cell Sci. 2016;129(24):4466-79. https://doi.org/10.1242/jcs.196717.
10.Hermjakob H, Montecchi-Palazzi L, Lewington C, Mudali S, Kerrien S, Orchard S, et al. IntAct: an open source molecular interaction database. Nucleic Acids Res. 2004;32(Database issue):D452-5. https://doi.org/10.1093/nar/gkh052.
11.Li J, Liu C, Müller U, Zhao B. RIPOR2-mediated autophagy dysfunction is critical for aminoglycoside-induced hearing loss. Dev Cell. 2022 Sep 26;57(18):2204-20.e6. https://doi.org/10.1016/j.devcel.2022.08.011.
12.Bischoff AM, Huygen PL, Kemperman MH, Pennings RJ, Bom SJ, Verhagen WI, et al. Vestibular deterioration precedes hearing deterioration in the P51S COCH mutation (DFNA9): an analysis in 74 mutation carriers. Otol Neurotol. 2005 Sep;26(5):918-25. https://doi.org/10.1097/01.mao.0000185048.84641.e3.
13.Alonso AV, Aguado RG, Camerano AM, Enseñat JF, de la Fuente EO, Angulo CM. Hearing and vestibular impairment related to a variant (c.263G>C) of the COCH gene. Otolaryngol Head Neck Surg. 2024 Dec 12. https://doi.org/10.1002/ohn.1074. Epub ahead of print.
14.International Organization for Standardization (ISO 7029:2017). Edition 3, 2017 [Internet]. Available from: https://www.iso.org/standard/42916.html
15.Degree of hearing loss. American Speech-Language-Hearing Association [Internet]. [cited 2024 Aug 14]. Available from: https://www.asha.org/public/hearing/degree-of-hearing-loss
16.Li MM, Tayoun AA, DiStefano M, et al. ACMG Professional Practice and Guidelines Committee. Clinical evaluation and etiologic diagnosis of hearing loss: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2022;24(7):1392-406. https://doi.org/10.1016/j.gim.2022.03.018.
17.González-Aguado R, Gallo-Terán J, Onecha E, Morales-Angulo C. Cochleo-vestibular phenotype in patients with pathogenic variations in the ACTG1 gene. Acta Otorrinolaringol Esp. 2024. In press.
18.Kim SY, Kim BJ, Oh DY, et al. Improving genetic diagnosis by disease-specific, ACMG/AMP variant interpretation guidelines for hearing loss. Sci Rep. 2022;12(1):12457. https://doi.org/10.1038/s41598-022-16661-x.
19.Usami S, Nishio S. The genetic etiology of hearing loss in Japan revealed by the social health insurance-based genetic testing of 10K patients. Hum Genet. 2022;141(5-6):665-81. https://doi.org/10.1007/s00439-021-02371-3.
20.Aliazami F, Gilani S, Farhud D, Naraghi M, Afshari M, Eslami M. Epidemiology, etiology, genetic variants in non-syndromic hearing loss in Iran: A systematic review and meta-analysis. Int J Pediatr Otorhinolaryngol. 2023;168:111512. https://doi.org/10.1016/j.ijporl.2023.111512.
21.Reda del Barrio S, García Fernández A, Quesada-Espinosa JF, et al. Genetic diagnosis of childhood sensorineural hearing loss. Acta Otorrinolaringol Esp. 2024;75(2):83-93. https://doi.org/10.1016/j.otoeng.2023.09.005.
22.Reda Del Barrio S, de Vergas Gutiérrez J, Quesada-Espinosa JF, et al. Diagnostic yield of genetic testing in adults with sensorineural hearing loss. Acta Otorrinolaringol Esp. 2024;10:S2173-5735(24)00026-7. https://doi.org/10.1016/j.otoeng.2023.10.007.
23.Cabanillas R, Diñeiro M, Cifuentes GA, et al. Comprehensive genomic diagnosis of nonsyndromic and syndromic hereditary hearing loss in Spanish patients. BMC Med Genomics. 2018;11:58. https://doi.org/10.1186/s12920-018-0375-5.
24.Del Castillo I, Morín M, Domínguez Ruiz M, Moreno Pelayo MA. Genetic etiology of non-syndromic hearing loss in Europe. Hum Genet. 2022;141(5-6):683-96. https://doi.org/10.1007/s00439-021-02425-6.
25.Seligman KL, Shearer AL, Frees K, Nishimura C, Kolbe D, Dunn C, et al. Genetic causes of hearing loss in a large cohort of cochlear implant recipients. Otolaryngol Head Neck Surg. 2022;166(4):734-7. https://doi.org/10.1177/01945998211021308.